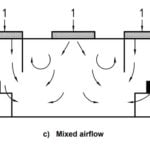
Some argue new PMA (pre-market approval) for e-liquid manufacturers and some retailers unfairly penalize smaller e-liquid manufacturing organizations. Others have urged the FDA for more strict market controls, suggesting that manufacturers have not prioritized consumer safety or established adequate manufacturing processes. While most large scale production facilities have standardized manufacturing controls, those mixing liquids in small facilities without environmental controls may struggle to receive pre-market approval. Based on FDA foreshadowing, most e-vape and liquid manufacturers will require some type of cleanroom or clean area in the near future. This post clarifies and outlines the FDA roadmap and current guidelines for E-liquid production.
Future FDA Regulations for E-Liquids and Electronic Tobacco Products
We’ve compiled the most relevant documentation on the FDA’s scope for e-liquid facilities, which is currently guided by Good Manufacturing Practices (GMP) and local/federal guidelines.
FDA E-Liquid Requirements
We reached out to the FDA on its requirements for e-liquid cleanrooms and manufacturing environments. Our interpretation of their response indicates looming regulation for control methods and environmental standards during ENDS production. In most FDA facilities, the guidance combines applicable standards established by GMP, USP, ISO, AEMSA, OSHA, NIOSH, and state-commissioned oversight boards.
The FDA’s generalized statement is as follows:
Thank you for contacting the U.S. Food and Drug Administration’s (FDA or the Agency) Center for Tobacco Products (CTP).
The Federal Food, Drug, and Cosmetic Act (FD&C Act or the Act) gives FDA the authority to regulate the manufacturing, marketing, sale, and distribution of tobacco products in the United States. Section 201(rr)(1) of the FD&C Act defines “tobacco product” as “any product made or derived from tobacco that is intended for human consumption, including any component, part or accessory of a tobacco product.” FDA regulates cigarettes, cigarette tobacco, roll-your-own tobacco, and smokeless tobacco. The Deeming rule, which published in the Federal Register on May 10, 2016, and took effect on August 8, 2016, extended FDA’s authority to deemed tobacco products such as electronic cigarettes, cigars, hookah, and pipe tobacco, as well as their components and parts, but not their accessories.
Component or part means any software or assembly of materials intended or reasonably expected to alter or affect the tobacco product’s performance, composition, constituents, or characteristics, or to be used with or for the human consumption of a tobacco product.
FDA must first promulgate through rulemaking good manufacturing practice requirements applicable to tobacco products (or Tobacco Product Manufacturing Practice). Once these requirements are in effect, they will apply to tobacco products.
One could understand forward-looking statements as an indication of regulatory foreshadow, The FDA is currently trying to understand the full scope of how many manufacturing facilities exist and the size and scope of each entity.
Other federal, state, or local requirements may apply to you and your products. For more information, please visit our website www.fda.gov/tobacco-products. We also encourage you to subscribe to FDA’s “CTP News” and “CTP Connect” newsletters. By subscribing, you’ll receive updates about regulatory activities, retailer notices, upcoming events, and public education campaigns.
“It would not protect the public health to forego implementation of these provisions until the FDA can issue final product standards and tobacco product manufacturing practice regulations. It is also important to note that this final deeming rule is a foundational rule that enables FDA to issue future regulations if FDA determines that they would be appropriate for the protection of public health.”
The FDA calculates the risks of additives and substances based on two questions: The first being, how much of that substance will a user consume? The second, how much of that substance is considered safe to consume based on scientific analysis? Based on these findings, the FDA builds complex regulatory standards, policies, restrictions, and, and manufacturing standards. Until the FDA fully understands the landscape and size of the market, it will not issue compliance standards for manufacturing.
Read More: What are the American E-Liquid Manufacturing Standards & Requirements?
Is E-Liquid FDA Approved?
Tobacco-related product manufacturers are subject to FDA pre-market approval.
Are Vape Shops Considered Manufacturers?
“Any person, including any repacker or relabeler, who (A) manufactures, fabricates, assembles, processes, or labels a tobacco product; or (B) imports a finished tobacco product for sale or distribution in the United States.”
“You are considered a manufacturer if you, for example, mix or prepare e-liquids, create or modify aerosolizing apparatus, repackage ENDS products, or relabel ENDS products atomizers, tanks, and accessories.”
21 CFR Part 1140 – CIGARETTES, SMOKELESS TOBACCO, AND COVERED TOBACCO PRODUCTS
- Subpart A – General Provisions (§§ 1140.1 – 1140.3)
- Subpart B – Prohibition of Sale and Distribution to Minors (§§ 1140.10 – 1140.16)
- Subpart C [Reserved]
- Subpart D – Labeling and Advertising (§§ 1140.30 – 1140.34)
The FDA’s summarized requirements for e-liquid manufacturers as of July 2019:
“The Federal Food, Drug, and Cosmetic Act (FD&C Act or the Act) gives FDA the authority to regulate the manufacturing, marketing, sale, and distribution of tobacco products in the United States. Section 201(rr)(1) of the FD&C Act defines “tobacco product” as “any product made or derived from tobacco that is intended for human consumption, including any component, part or accessory of a tobacco product.” FDA regulates cigarettes, cigarette tobacco, roll-your-own tobacco, and smokeless tobacco.”
“…FDA must first promulgate through rulemaking good manufacturing practice requirements applicable to tobacco products (or Tobacco Product Manufacturing Practice). Once these requirements are in effect, they will apply to tobacco products”
What Will Premarket Review of New Tobacco Products Require?
“Section 910(b) of the FD&C Act states that a PMTA shall contain full reports of all investigations of health risks; a full statement of all components, ingredients, additives, and properties, and of the principle or principles of operation of such tobacco product; a full description of methods of manufacturing and processing (which includes; a listing of all manufacturing, packaging, and control sites for the product); an explanation of how the product complies with applicable tobacco product standards; samples of the product and its components; and labeling.” PG. 432
Do I need Pre Market Approval?
What are the activities a vape shop can do without having to
1) submit applications for premarket review
2) register and list with the FDA
3) submit health documents
4) report ingredient lists or
5) report harmful and potentially harmful constituents? FDA E-Liquid FAQ #6
This guidance explains that FDA does not consider certain activities performed by vape shops to modify the tobacco product and, consequently, vape shops that perform these activities are not required to obtain premarket authorization for their products. In addition, FDA does not intend to enforce the other four previously mentioned requirements for these vape shops. Examples of these activities include:
- Demonstrating or explaining the use of an ENDS product without assembling the product;
- Maintaining an ENDS product by cleaning or tightening fixtures (e.g., screws);
- Replacing coils in an ENDS product with identical coils (e.g., same ohm and wattage rating); and
- Assembling a final product from the components and parts packaged together in an ENDS kit.
This guidance also explains that FDA does not intend to enforce the five requirements listed above for these vape shops if..
1. generally speaking, all modifications are consistent with the conditions of the FDA marketing authorization (MA) or
2.if the original manufacturer provides specifications and all modifications made are consistent with those specifications. Examples of these scenarios include:
- Refilling an open system ENDS if no further modifications are made to the device or to the e-liquid before, during or after the refill that are outside the FDA marketing authorization (MA) order;
- Refilling an open system ENDS if no further modifications are made to the device or the e-liquid before, during or after the refill that – if there is no MA order – are inconsistent with the manufacturer’s specifications.
What are the activities that, if performed, would trigger a retailer to become a manufacturer and, therefore, be required to comply with the requirements to 1) submit applications for premarket review 2) register and list with the FDA 3) submit health documents 4) report ingredient lists or 5) report harmful and potentially harmful constituents? FDA E- Liquid FAQ #7
Examples of activities that are considered modifying a tobacco product under the FD&C Act – and would thus require compliance with all five requirements listed above – include:
- Modifying a product outside of a FDA marketing authorization (MA) order;
- Refilling a closed system ENDS;
- Repairing and modifying a part (an atomizer head, for example) outside the conditions of an FDA marketing authorization (MA) order;
- Replacing a part (a coil in an ENDS product, for example) – that was on the market as of August 8, 2016 but that does not have an MA order – with a different part (for example, a coil that has a different ohm or wattage rating from that used by the original manufacturer); and
- Assembling a custom final product.
What if I Don’t Have a Listable Manufacturing Site?
Those considering an E-liquid manufacturing environment should consider the pros and cons of a building a cleanroom. In nearly every FDA regulated industry, “cleanroom” establish minimum standards for hygiene and quality enhancement.
Inspection failures, audits, and late evolving construction have negative impacts on product throughput, product quality, For example, a facility might need to halt all operations in the cases of failed inspections, audits, compliance revisions, re-audits, and publicly available FDA warnings.
Read More: What are the Requirements for E-Liquid Manufacturing Labs?
Will All Vape Shops Fall Under FDA Regulations?
Vape shops and convenience stores often sell liquids, nicotine, flavors, and other novel formulas, pre-mixed batches, and bulk supplies. Any of the above, particularly products packaged or combined on-location, are subject to strict control measures. Even retail locations, such as retail vape shops, fall within the definition of an ENDS system.
“You are considered a retailer if you sell tobacco products to individuals for personal consumption. Depending on the activities a vape shop engages in, it can be a tobacco product retailer, a tobacco product manufacturer, or both. Retailers and manufacturers are subject to inspection by FDA.” – FDA
Regardless of the point of sale, whether online or on-location, nearly every for-sale item related to e-cigarettes, vapes, and e-liquid products fall under this premise.
Not understated, is the importance of intermediaries, distributors, retailers, and services in the final delivery of a quality-controlled device or liquid to the end-user. The FDA deems these as examples of Electronic Nicotine Delivery Systems (ENDS).
Chronology of FDA E-Liquid and E-Cigarettes Manufacturing Statements
This chronology is considered a snapshot of FDA press releases and publically available documentation, it is by no means exhaustive and subject to future updates.
2014
The beginnings of the FDA stance on E-cigarette begins with Federal Register / Vol. 79 , No. 80 21 CFR Parts 1100, 1140, and 1143 issued Friday, April 25, 2014. It states:
“Components and parts that would be covered under this proposal include those items sold separately or as part of kits sold or distributed for consumer use or further manufacturing or included as part of a finished tobacco product. Such examples would include air/smoke filters, tubes, papers, pouches, or flavorings used for any of the proposed deemed tobacco products (such as flavored hookah charcoals and hookah flavor enhancers) or cartridges for e-cigarettes.”
2016
This documentation is continued with Federal Register Volume 81, No, 90 issued May 10, 2016. This rule is dated as effective August 8, 2016:
“The Deeming rule, which published in the Federal Register on May 10, 2016, and took effect on August 8, 2016, extended FDA’s authority to deemed tobacco products such as electronic cigarettes, cigars, hookah, and pipe tobacco, as well as their components and parts, but not their accessories. Component or part means any software or assembly of materials intended or reasonably expected to alter or affect the tobacco product’s performance, composition, constituents, or characteristics, or to be used with or for the human consumption of a tobacco product.”
“As of the effective date, persons who own or operate a domestic establishment engaged in the manufacture, preparation, compounding, or processing of tobacco products (hereinafter, ‘‘manufacturing establishments’’) will be subject to the registration requirements. FDA will thus receive information on the location and number of manufacturing establishments, which will allow the Agency to establish effective compliance programs.”
“However, the FDA may request additional information about your PMTA as necessary. FDA may also want to inspect your manufacturing, clinical research, or nonclinical research sites to support its review of your PMTA. Inspections of these sites allow FDA to assess the accuracy and validity of the information provided, including clinical and non-clinical information, and confirm that the product can be manufactured in the way that the PMTA specifies. Inspections will also provide important information regarding whether the manufacturing, processing, or packing of the tobacco product conforms to tobacco product manufacturing practices, which will be set forth in a future rulemaking. (Premarket Tobacco Product Applications for Electronic Nicotine Delivery Systems – Pg. 12) [2016]
2019
We can look further ahead in the timeline by the FDA guidance on achieving premarket tobacco approval (PMTA) for e-liquid manufacturers.
July 15th 2019:
“U.S. District Court judge in Maryland issued a decision that, among other things, requires makers and importers of e-cigarettes and other electronic nicotine delivery systems (ENDS) and certain other tobacco products like cigars, pipe tobacco and hookah to submit applications for their currently marketed products to the agency within 10 months.” FDA Statement
“As explained in the deeming rule, the FD&C Act authorizes FDA to regulate the manufacture of new tobacco products including those manufactured at the retail level. This is important to FDA’s ability to protect the public health because products manufactured at the retail level pose many of the same risks as those manufactured upstream and possibly additional risks related to the lack of standard manufacturing practices and controls (81 FR 28973 at 29044)”
“FDA intends to issue regulations under section 906(e) that will contain the requirements for tobacco product manufacturing practices. At that time, each new PMTA will also be expected to demonstrate that the methods, facilities, or controls used conform to these regulations (section 910(c)(2)(B))”
August 2nd, 2019
August 8th, 2019:
FDA Regulation and Enforcement
The FDA issues warnings to manufacturers in the form of a written letter which is made available to the public online. It’s the manufacturer’s responsibility to respond within 14 days providing evidence, reasoning, justification, or exemption requests.
Warning Letters – are sent to the individuals or firms, advising them of specifically noted violations. These letters request a written response as to the steps which will be taken to correct the violation. These letters constitute one form of warning that can be issued under the current Agency policy.
In the case that techniques, processes, or equipment do not comply with federal or state inspection requirements, facilities must take appropriate action:
- Revising SOP with protocol and procedures that remedy deficiencies
- Obtaining invoices for required supplies
- Create training materials and records for corrective actions
- Observe revised procedure in action
Failure to remedy, reverse, or argue exemption leaves three possible scenarios:
Seizure – An action brought against an FDA-regulated product because it is adulterated and/or misbranded within the meaning of the Act. The purpose of such an action is to remove specific violative goods from commerce.
Injunction – An order by a court that requires an individual or corporation to do or refrain from doing a specific act. FDA may seek injunctions against individuals and/or corporations to prevent them from violating or causing violations of the Act.
Criminal prosecution – may be recommended in appropriate cases for violation of Section 301 of the Act.; Misdemeanor convictions, which do not require proof of intent to violate the Act, can result in fines and/or imprisonment up to one year. Felony convictions, which apply in the case of a second violation or intent to defraud or mislead, can result in fines and/or imprisonment up to three years.
Criminal Fines for Food Drug and Cosmetic Act Violations
Misdemeanor fines under the Act may reach $500,000 under some circumstances. The Criminal Fine Enforcement Act of 1994 (Public Law 98-596) provides for fines for violations of Federal law. Although it is not part of the Act, the Criminal Fine Enforcement Act of 1994 applies to all fines levied under the Act, as well as other statutes that contain provisions enforced by FDA.
The following fines are applicable for each offense:
Up to $100,000 for a misdemeanor by an individual that does not result in death.
Up to $200,000 for a misdemeanor by a corporation that does not result in death.
Up to $250,000 for a misdemeanor by an individual that results in death, or a felony.
Up to $500,000 for a misdemeanor by a corporation that results in death, or a felony.
The type of action recommended will depend upon the nature of the violation and the public health concern, Agency policy, previous history of violations by the firm, and other factors.
FDA Contact Information for Small Businesses and Tobacco Products
For any specific questions with regards to your current or future business, the FDA’s contact information for small business inquiries is as follows:
Contact the FDA by Mail:
Food and Drug Administration
Center for Tobacco Products
10903 New Hampshire Avenue
Silver Spring, MD 20993
Contact the FDA by Email: [email protected]
Contact the FDA by Phone: 1-877-287-1373
Advantages of Cleanrooms for E-Liquid Manufacturing and FDA Regulated Industries
Controlled environments get products to market faster with fewer variables, especially under the watchful eye of the FDA. In decades past, a customized cleanroom build required hundreds of thousands of dollars. Today, there are a number of cleanroom systems available for just a few thousand dollars. Nearly every manufacturer or mixing/packaging retailer uses some type of cleanroom or clean area. In non-regulated industries, cleanroom workflows and environmental controls subdue aerosol and surface contamination.
Cleanroom technology is adopted in nearly every industry regulated by the FDA, but also in non-regulated industries. Why would companies deploy cleanrooms if they didn’t have to by law? It’s simple — Because clean, consistent products with batch-to-batch consistency, low defect rates, and the longest possible shelf life are more profitable.
The area could consist of a temporary softwall curtain structure or a sprawling hardwall facility for mass assembly and packaging. In retail environments, a small containment area is often favored for custom e-juice mixes and preparing other batch ingredients for final packing. Some require large-scale environments for high-volume, and often automated filling, packaging, and bottling lines.