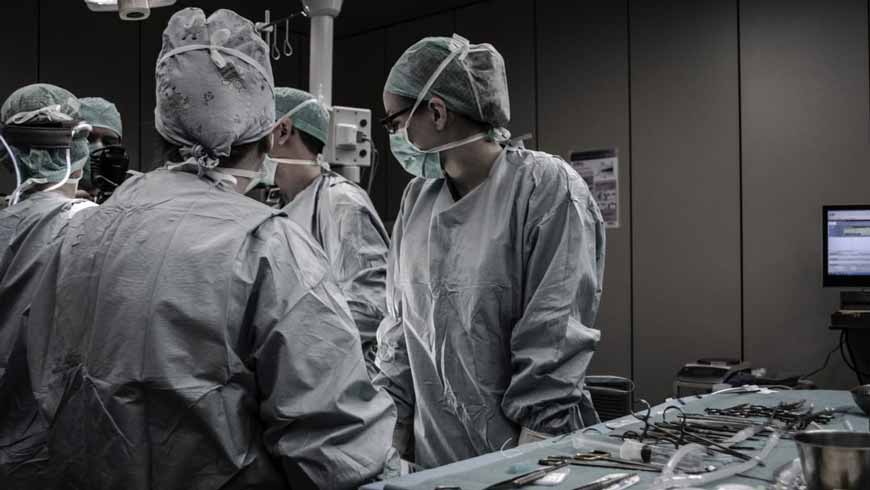
Class 2 - 3 Medical Device Cleanrooms
This post classifies cleanroom design parameters during medical device manufacturing. Establishing criteria is a challenge. The FDA does not set forth any cleanroom classification or particulate level based on application.
Essential Medical Devices Require Cleanroom Production
All invasive procedures involve contact between a medical device or surgical instrument and a patient’s sterile tissue or mucous membranes. A major risk of all such procedures is the introduction of pathogenic microbes that could lead to infection. Failure to properly disinfect or sterilize new or reusable medical equipment is a perilous risk in the case of a breached sterile barrier.
Environmental controls are dependent on the intended use of an object: items that contact sterile tissues are considered critical items such as surgical instruments or implantable devices. Semicritical items which contact mucous membranes including endoscopes in medical fields or even drill guides for dental work. Items that only contact skin are considered noncritical items, such as stethoscopes. Each of these categories designates a different threshold of sterilization and disinfection which include high-level disinfection and low-level disinfection.
ISO 14644 - FS-209E Medical Device Cleanrooms
Authoritative documents such as ISO 14644 and FS-209E provide no specific instructions regarding activities carried out in medical device environments. Broadly, medical device manufacturing is conducted in an ISO 5 – 8 cleanroom (Class 100 – 100,000). Medical device packaging is conducted in an ISO Class 7- 8 cleanroom. An ISO 8 environment may include a dedicated gowning room and sometimes softwall environments in packaging or preparation areas. Classification of surrounding environments, isolators, and sterile barriers all require very specific performance criteria.
Related Post: USP 797 Sterile Compounding
The above standards do not apply expectations of the state board or FDA mandates. ISO classifications simply standardize a benchmark of environmental quality.
Recently expanded guidelines, ISO 13485 (medical devices) and USP 800 (pharmaceutical compounding), emphasize an evolving landscape of risk management solutions for medical sciences. Harmonizing medical device cleanroom standards requires an evaluation of not only cleanroom design, but also processes equipment, materials, consumables, and assemblies.
FDA Medical Devices in Plain Language (Summarized)
Medical devices are defined in Section 201(h) of the Food, Drug, and Cosmetic Act as any healthcare product that does not require chemical action or metabolization.
A medical device is an instrument, apparatus, implement, machine, contrivance implant, in-vitro reagent, or other similar or related article including any component, part or accessory. The FDA classifies medical devices in over 1700 generic device categories within 16 medical specialties. Each classification outlines a unique compliance based on risk estimates.
(1) A medical device is recognized in the official National Formulary, or the US Pharmacopeia, or Formulary, or the US Pharmacopeia, or any supplement to them.
(2) A medical device is intended for use in the diagnosis or cure of disease or other conditions including the cure, mitigation, treatment, or prevention of mitigation, or prevention of disease, in man or other animals in man or other animals, or…
3) A medical device does not achieve its primary intended purposes through chemical action or metabolization within or on the body of a man or other animals.
Medical Devices Classes and Quality Management
Class 1 devices are perceived to be the lowest risk devices. A formal review (i.e., 510(k) or PMA) is not typically required. General Controls are usually sufficient. Approximately ¾ of all devices listed with the FDA each year are Class 1. Examples include medical tape and bandages.
Class 2 devices are perceived to be of greater risk to the patient or user. General controls are not sufficient for safety or efficacy. Most of these devices require a 510(k) application. Approximately 3,000 Class 2 devices are cleared by the FDA each year. Most coronary catheters fall within this classification.
Class 3 devices are perceived to represent the highest risk to patient or user. These devices support or sustain human life, may prevent or impair human health, or present potential risk of illness or injury. Most Class 3 devices require a PMA application. Approximately 40 Class 3 devices are approved by the FDA each year. Examples include heart valves and pacemakers.
In regards to medical device cleanroom design, Class II and Class III devices require a Quality Management System established by ISO 13485. A cleanroom is a critical component for ensuring traceability, lot-to-lot tracking, establishing an aseptic workflow, and for monitoring air quality to identify and eliminate sources of contamination.
Related Post: ISO 13485 Quality Management Systems
The requirements of ISO 13485 apply to any medical device organization regardless of size and activity as a basis for demonstrating and supporting compliance with regulatory requirements.
Medical Device Cleanroom Design
The principles of cleanroom design for medical devices must support three objectives:
- Suitable size, construction, and location to facilitate cleaning, maintenance, and proper operations
- Plan adequate space for orderly placement of equipment and materials to prevent mix-ups and contamination
- Design the adequate flow of materials & persons to prevent contamination.
FDA 21 CFR 211.42 addresses design and construction features. According to the regulation:
- The facility must be of suitable size, construction, and location so that cleaning, maintenance, and operations can be properly performed.
- The facility must have adequate space and flow design to prevent mix-ups and cross-contamination.
- Surfaces must be easy to clean.
- Temperature and humidity must be controlled as appropriate.
- Air must be positive pressure and HEPA filtered. (Non-hazardous)
- Environmental conditions must be monitored.
- Room and equipment must be cleaned and disinfected properly.
- Equipment must remain in aseptic conditions.
Medical Device Cleanroom Requirements?
What Cleanroom Classification is Required for Class I, Class II, and Class III Devices
Our cleanroom teams often field inquiries from large and small operations alike. Many of our customers want to know what ISO Classification is required for a Class I or Class II device, or for a particular technology. Some organizations don’t require a standardized compliance and only intend to make a particular area cleaner, or create manufacturing cells that isolate cleaner tasks from dirtier ones.
ISO 7 – 8 cleanrooms are common for Class I and Class II medical devices. Because a Class 1 device does not require a Quality Management System, a softwall cleanroom is common instead of a dedicated installation. Class III devices carry greater risk and a higher degree of regulatory control, therefore justify the most stringent manufacturing conditions. As device complexity grows and part sizes shrink, particles become more problematic. This leads many facilities towards a cleaner classification than immediately necessary in order to future-proof the build for evolving process requirements.
Part 1: Medical Device Cleanroom Walls, Windows, & Containment
Part 2: Medical Device Cleanroom Doors, Electrical, & Partitions
Part 3: Medical Device Cleanroom Fan Filter Units – Ceiling Grids
Part 4: Medical Device Cleanroom Flooring
Part 5: Medical Device Cleanroom HVAC Design & Fan Filter Integration
Part 6: Pressure Differentials, Humidity and Temperature Calibration
Cleanroom Design for Injection Molded Medical Devices
Broadly, most injection molded medical devices are produced in ISO Class 8 (Class 100,000) cleanrooms. Sterile devices may require an ISO Class 5 cleanroom (Class 100), while medical device packaging is conducted in an ISO Class 7-8 cleanroom. Classification of surrounding environments, isolators, and sterilization processes all require very specific performance criteria.
Plastic injection molding and packaging may extend to ISO Class 8 (Class 100,000) which requires an average airflow velocity of 8 ft/min with 5 – 48 air changes per hour. The space will require HEPA filtration fan filter units with 5% – 20% ceiling surface coverage depending on the process.
Want More Info on a Cleanroom for Your Specific Application?
We help facilities produce yields faster with better process control. Get help identifying the proper equipment and cleanroom design for your facility.

Related Posts
 Medical Device Cleanroom Construction Part 1: Walls and Installation
Medical Device Cleanroom Construction Part 1: Walls and InstallationHow is a medical device cleanroom built? Here's a component-by-component guide to ISO Class 7 cleanroom construction.
 Medical Device Cleanroom Construction Part 2: Doors, Electrical, and Layout
Medical Device Cleanroom Construction Part 2: Doors, Electrical, and LayoutIn medical device cleanrooms, a common configuration is an ISO 8 gowning room and ISO 7 primary production area. A cascading air flow design and positive pressure HVAC calibration move the cleanest air at the…
 Medical Device Cleanroom Construction Part 3: Fan Filter Units - Ceiling Grids
Medical Device Cleanroom Construction Part 3: Fan Filter Units - Ceiling GridsIn this post, we overview technical considerations for clean room fan filter unit selection, installation, and features. You'll learn how PAC approaches design and configuration of cleanroom HEPA filter units and fan filter housing specifications.…
 Medical Device Cleanroom Construction Part 4 - VCT Flooring Design
Medical Device Cleanroom Construction Part 4 - VCT Flooring DesignThere are a few final preparations required before the final cleanroom flooring installation. Here's what to expect and consider for cleanroom tile installations and flooring installation.
 Medical Device Cleanroom Construction Part 5: HVAC Balancing & Calibration
Medical Device Cleanroom Construction Part 5: HVAC Balancing & CalibrationBefore cleanroom validation, a cleanroom requires mechanical calibration. HVAC calibration first ensures that the room meets the performance parameters which would deliver the expected results during final ISO validation, such as differential pressure, air change…
 Medical Device Cleanroom Construction Part 6: Pressure Differentials, Humidity and Temperature Calibration
Medical Device Cleanroom Construction Part 6: Pressure Differentials, Humidity and Temperature CalibrationThe central controller integrates the data from both the thermostats, fan filter processors, and room-side environmental sensors. Fan filter speed is then adjust by each group to achieve the specified room pressure while at ideal…
 ISO 13485:2016 Medical Devices
ISO 13485:2016 Medical DevicesISO 13485 is designed for use throughout the life cycle of a medical device. It supports each stage of medical device development and operation from initial concept to production and disposal. The standard helps internal…
 Cleanroom Classification Comparison Specifications Defined
Cleanroom Classification Comparison Specifications DefinedA comparison of cleanroom classifications and specifications.
 CleanPro® Softwall Cleanroom Enclosure
CleanPro® Softwall Cleanroom EnclosureThis customer needed to enclose a piece of machinery, and CleanPro® was able to provide a solution.
 CleanPro® Turnkey Cleanroom Install
CleanPro® Turnkey Cleanroom InstallCleanPro’s turn-key cleanroom solution provided a one-stop, one-contact result for the initial delivery and on-site installation of walls, ceiling grids, electrical systems, flooring, filters, HVAC, and more.
 Cleanroom Construction FAQ
Cleanroom Construction FAQDefinitive Cleanroom Construction FAQ: How much does a cleanroom cost? How does the level of ISO classification impact build and operating costs? How much supporting space will I need for a cleanroom addition or ISO…
 CleanPro® Chemotherapy & CSP Cleanroom Installation
CleanPro® Chemotherapy & CSP Cleanroom InstallationThis modular, sterile compounding cleanroom is designed for USP 797 and USP 800 compliance, particularly for compounding chemotherapy drugs. Safe handling of sterile compounds requires special considerations: heat-welded floors, anterooms and buffer areas.
- Cleanroom Construction FAQ
Definitive Cleanroom Construction FAQ: How much does a cleanroom cost? How does the level of ISO classification impact build and operating costs? How much supporting space will I need for a cleanroom addition or ISO…



